

Słowa kluczowe
DFT calculations
benzocarbazoles
Marcus-Hush theory of electron transport
Jak cytować
Abstrakt
W artykule przedstawiono wyniki obliczeń kwantowo-mechanicznych mających na celu poszukiwanie cząsteczki organicznego półprzewodnika charakteryzującego się niską energią transferu ładunku. Obliczenia z wykorzystaniem teorii funkcjonału gęstości (DFT) zostały wykonane dla karbazolu (Cz) i trzech izomerów benzokarbazolu, benzo(a)karbazolu (BaCz) , benzo(b)karbazolu (BbCz) i benzo(c)karbazolu (BcCz) dowodzą , że można uniknąć wzrostu energii reorganizacji cząsteczki tak dla przewodnictwa dziurowego jak i dla przewodnictwa elektronowego pomimo zwiększenia rozmiarów przestrzennych szkieletu aromatycznego cząsteczki. Cząsteczki benzo(b)karbazolu o największym rozmiarze podłużnym szkieletu d = 9,05 Å charakteryzują się najniższą energią reorganizacji dla przewodnictwa dziur 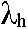 = 0,18 eV oraz najniższą wśród izomerów benzokarbazolu energią reorganizacji dla przewodnictwa elektronowego
= 0,18 eV oraz najniższą wśród izomerów benzokarbazolu energią reorganizacji dla przewodnictwa elektronowego 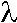 e= 0,11 eV. Analiza odległości pomiędzy wybranymi charakterystycznymi atomami wodoru pozwala przypuszczać, że zmniejszenie energi reorganizacji obserwowane dla benzo(b)- karbazolu jest związane ze zmiejszaniem wewnątrzcząsteczkowych oddziaływań pomiędzy sąsiadującymi atomami wodoru.
e= 0,11 eV. Analiza odległości pomiędzy wybranymi charakterystycznymi atomami wodoru pozwala przypuszczać, że zmniejszenie energi reorganizacji obserwowane dla benzo(b)- karbazolu jest związane ze zmiejszaniem wewnątrzcząsteczkowych oddziaływań pomiędzy sąsiadującymi atomami wodoru.
Bibliografia
Varathan E., Vijayc D., Subramanian V. 2016. Quantum chemical design of carbazole-and pyridoindole-based ambipolar host materials for blue phosphorescent OLEDs. RSC Adv. 6: 74769-74783. DOI: 10.1039/c6ra15748c
Marzinzik A.L., Rademacher P., Zander M. 1996. Structural chemistry of polycyclic heteroaromatic compounds. Part 9. Photoelectron spectra and electronic structures of anellated carbazoles. Extension of Clar's aromatic sextet model to hetarenes. J. Mol. Struct. 375: 117-126. https://doi.org/10.1016/0022-2860(95)09029-0
Batra I.P., Bagus P.S., Clementi E., Seki H. 1973. Ab initio Calculations for the Electronic Structure of Carbazole and Trinitrofluorenone. Theoret.Chim. Acta (Berl.) 32: 279-293. https://link.springer.com/article/10.1007/BF00526864
Gajda K., Zarychta B., Kopka K., Daszkiewicz Z., Ejsmont K. 2014. Substituent effects in nitro derivatives of carbazoles investigated by comparison of lowtemperature crystallographic studies with density functional theory (DFT) calculations. Acta Cryst. C70: 987–991. doi:10.1107/S205322961402634 A DFT study of reorganization energy of carbazole derivatives
Gaussian 09, Revision A.02. 2009. Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., M. Ehara, Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A., Peralta Jr., J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam J. M., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas O., Foresman J. B., Ortiz J. V., Cioslowski J., Fox D. J., Wallingford CT: Gaussian, Inc.
Becke, J. 1996. Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing. Chem. Phys. 104: 1040-1046. https://doi.org/10.1063/1.470829
Lee C., Yang W., Parr R.G. 1998. Development of the Colle-Salvetti correlationenergy formula into a functional of the electron density. Phys. Rev. B 37: 785-789. https://doi.org/10.1103/PhysRevB.37.785
Pan J-H., Chiu H-L., Wang B-C. 2005. Theoretical investigation of carbazole derivatives as hole-transporting materials in OLEDs. Journal of Molecular Structure: THEOCHEM 725: 89–95. doi:10.1016/j.theochem.2005.02.061
Hlel A., Mabrouk A., Chemek M., Ben Khalifa I., Alimi K. 2014. A DFT study of charge-transfer and opto-electronic properties of some new materials involving carbazole units. Computational Condensed Matter 3: 30-40. http://dx.doi.org/ 10.1016/j.cocom.2015.02.001
Marcus R.A. 2000. Tutorial on rate constants and reorganization energies. J. Electroanalytical Chem. 483: 2-6. http://dx.doi.org/10.1016/S0022-0728(00)00011-5
Marcus, R.J. 1993. Electron transfer reactions in chemistry. Theory and experiment. Rev. mod. phys. 65: 3, 599-610. DOI:https://doi.org/10.1103/RevModPhys.65.599
Malloci G., Cappellini G., Mulas G., Mattoni A. 2011. Electronic and optical properties of families of polycyclic aromatic hydrocarbons: a systematic (timedependent) density functional theory study. Chem. Phys. 384(1): 19-27. https://doi.org/10.1016/j.chemphys.2011.04.013
Malloci G., Mulas G., Capellini G., Joblin C. 2007. Time-dependent density functional study of the electronic spectra of oligoacenes in the charge states .1, 0, +1, and +2, Chem. Phys. 340: 43-58. DOI: 10.1016/j.chemphys.2007.07.046
Tsuneda T., Song J.-W., Suzuki S., Hirao K. 2010. On Koopmans’ theorem in density functional theory. Chem. Phys. 133, 174101-1 -174101-9. https://doi.org/10. 1063/1.3491272